
Huntington-Krankheit erstmals erfolgreich behandelt

Eine der grausamsten und verheerendsten Krankheiten – die Huntington-Krankheit – wurde laut Ärzten erstmals erfolgreich behandelt.
Die Krankheit tritt familiär gehäuft auf, tötet unaufhaltsam Gehirnzellen und ähnelt einer Kombination aus Demenz, Parkinson und Motoneuron-Krankheit.
Ein emotionales Forschungsteam war den Tränen nahe, als es beschrieb, wie die Daten zeigten, dass sich der Krankheitsverlauf bei den Patienten um 75 % verlangsamte.
Das bedeutet, dass der Rückgang, den man normalerweise innerhalb eines Jahres erwarten würde, erst nach vier Jahren nach der Behandlung eintreten würde, was den Patienten Jahrzehnte „guter Lebensqualität“ beschert, sagte Prof. Sarah Tabrizi gegenüber BBC News.
Bei der neuen Behandlung handelt es sich um eine Art Gentherapie, die im Rahmen einer 12 bis 18 Stunden dauernden, heiklen Gehirnoperation durchgeführt wird.
Die ersten Symptome der Huntington-Krankheit treten meist im Alter zwischen 30 und 40 Jahren auf und führen normalerweise innerhalb von zwei Jahrzehnten zum Tod. Dies eröffnet die Möglichkeit, dass eine frühzeitigere Behandlung das Auftreten der Symptome von vornherein verhindern könnte.
Prof. Tabrizi, Direktor des Huntington-Krankheitszentrums des University College London, bezeichnete die Ergebnisse als „spektakulär“.
„Nicht einmal in unseren kühnsten Träumen hätten wir mit einer 75-prozentigen Verlangsamung des klinischen Verlaufs gerechnet“, sagte sie.
Keiner der behandelten Patienten wurde identifiziert, aber einer wurde aus medizinischen Gründen in den Ruhestand versetzt und ist wieder in den Beruf zurückgekehrt. Andere Studienteilnehmer können noch gehen, obwohl sie voraussichtlich einen Rollstuhl benötigen.
Die Behandlung dürfte sehr teuer sein. Dennoch ist dies ein Moment echter Hoffnung angesichts einer Krankheit, die Menschen in ihrer Blütezeit trifft und Familien zerstört.

Die Huntington-Krankheit ist in Jack May-Davis‘ Familie weit verbreitet. Er trägt das fehlerhafte Gen, das die Krankheit verursacht, ebenso wie sein Vater Fred und seine Großmutter Joyce.
Jack sagte, es sei „wirklich furchtbar und schrecklich“ gewesen, den unaufhaltsamen Verfall seines Vaters mitzuerleben.
Die ersten Symptome traten auf, als Fred Ende 30 war. Dazu gehörten Veränderungen in seinem Verhalten und seiner Bewegungsart. Schließlich benötigte er rund um die Uhr Palliativpflege, bevor er 2016 im Alter von 54 Jahren starb.
Jack ist 30, Rechtsanwaltsgehilfe, frisch mit Chloe verlobt und hat an einer Studie am UCL teilgenommen, um seine Diagnose ins Positive zu wenden.
Aber er wusste immer, dass er dazu bestimmt war, das Schicksal seines Vaters zu teilen, bis heute.
Jetzt sagt er, der „absolut unglaubliche“ Durchbruch habe ihn „überwältigt“ und er könne einer Zukunft entgegensehen, „die etwas rosiger erscheint. Sie lässt mich tatsächlich daran denken, dass mein Leben noch viel länger sein könnte.“

Die Huntington-Krankheit wird durch einen Fehler in einem Teil unserer DNA, dem sogenannten Huntingtin-Gen, verursacht.
Wenn einer Ihrer Elternteile an der Huntington-Krankheit leidet, besteht eine 50-prozentige Wahrscheinlichkeit, dass Sie das veränderte Gen erben und schließlich ebenfalls an der Huntington-Krankheit erkranken.
Diese Mutation macht ein normales, im Gehirn benötigtes Protein – das sogenannte Huntingtin-Protein – zu einem Neuronenkiller.
Ziel der Behandlung ist es, den Spiegel dieses toxischen Proteins mit einer einzigen Dosis dauerhaft zu senken.
Die Therapie nutzt modernste genetische Medizin, die Gentherapie und Gen-Silencing-Technologien kombiniert.
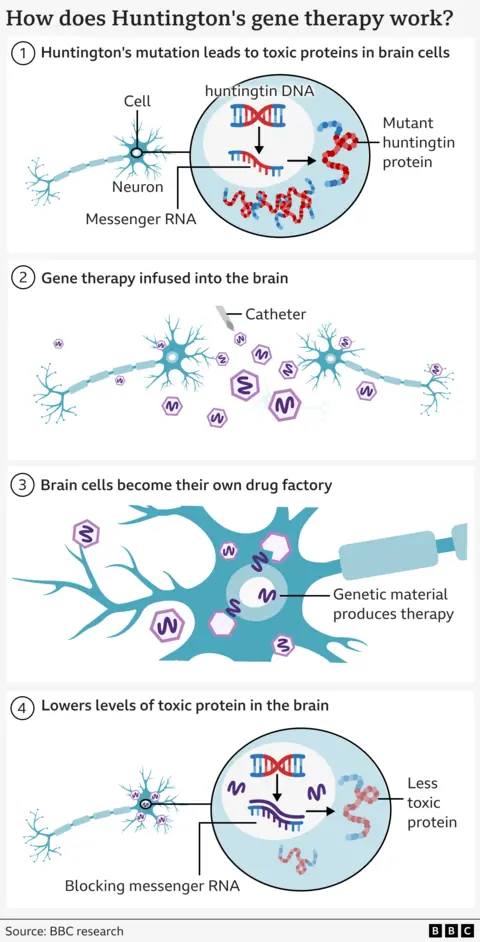
Es beginnt mit einem sicheren Virus, das so verändert wurde, dass es eine speziell entwickelte DNA-Sequenz enthält.
Dieses wird mithilfe einer Echtzeit-MRT-Untersuchung tief in das Gehirn infundiert, um einen Mikrokatheter zu zwei Hirnregionen zu führen – dem Nucleus caudatus und dem Putamen. Dieser neurochirurgische Eingriff dauert 12 bis 18 Stunden.
Das Virus fungiert dann als mikroskopischer Postbote: Es liefert das neue DNA-Stück in die Gehirnzellen, wo es aktiv wird.
Dadurch werden die Neuronen zu Fabriken, die die Therapie herstellen, um ihren eigenen Tod abzuwenden.
Die Zellen produzieren ein kleines Fragment genetischen Materials (sogenannte Mikro-RNA), das die von der DNA der Zellen gesendeten Anweisungen (sogenannte Messenger-RNA) zum Aufbau von mutiertem Huntingtin abfangen und deaktivieren soll.
Dies führt zu niedrigeren Konzentrationen des mutierten Huntingtins im Gehirn.
Die Ergebnisse der Studie, an der 29 Patienten teilnahmen, wurden in einer Erklärung des Unternehmens uniQure veröffentlicht, sind jedoch noch nicht vollständig zur Überprüfung durch andere Spezialisten veröffentlicht worden.
Die Daten zeigten, dass drei Jahre nach der Operation eine durchschnittliche Verlangsamung der Krankheit um 75 % zu verzeichnen war. Die Messung basiert auf einer Messung, die Kognition, Motorik und die Fähigkeit zur Bewältigung des täglichen Lebens berücksichtigt.
Die Daten zeigen auch, dass die Behandlung Gehirnzellen rettet. Der Gehalt an Neurofilamenten in der Rückenmarksflüssigkeit – ein deutliches Zeichen für das Absterben von Gehirnzellen – hätte bei einem weiteren Krankheitsverlauf um ein Drittel ansteigen müssen, war aber tatsächlich niedriger als zu Beginn der Studie.
„Das ist das Ergebnis, auf das wir gewartet haben“, sagte Prof. Ed Wild, beratender Neurologe am National Hospital for Neurology and Neurosurgery am UCLH.
„Es bestand durchaus die Möglichkeit, dass wir ein solches Ergebnis nie sehen würden. Da wir in einer Welt leben, in der wir wissen, dass dies nicht nur möglich ist, sondern dass das tatsächliche Ausmaß der Wirkung atemberaubend ist, ist es sehr schwierig, die Emotionen vollständig einzufangen.“
Er sagte, er sei „ein bisschen den Tränen nahe“, wenn er an die möglichen Auswirkungen auf die Familien denke.
Die Behandlung galt als sicher, obwohl bei einigen Patienten durch das Virus eine Entzündung auftrat, die Kopfschmerzen und Verwirrtheit verursachte, die entweder verschwanden oder eine Steroidbehandlung erforderlich machten.
Prof. Wild geht davon aus, dass die Therapie „lebenslang anhalten sollte“, da Gehirnzellen vom Körper nicht auf die gleiche Weise ersetzt werden wie Blut, Knochen und Haut ständig erneuert werden.
In Großbritannien, den USA und Europa leiden etwa 75.000 Menschen an der Huntington-Krankheit, Hunderttausende sind Träger der Mutation, was bedeutet, dass sie an der Krankheit erkranken werden.
UniQure will im ersten Quartal 2026 eine Zulassung in den USA beantragen und das Medikament noch im selben Jahr auf den Markt bringen. Gespräche mit den Behörden in Großbritannien und Europa beginnen im nächsten Jahr, der Fokus liegt zunächst jedoch auf den USA.
Dr. Walid Abi-Saab, der leitende Arzt bei uniQure, sagte, er sei „unglaublich gespannt“ auf die Bedeutung der Ergebnisse für die Familien und fügte hinzu, die Behandlung habe „das Potenzial, die Huntington-Krankheit grundlegend zu verändern“.
Aufgrund des hohen Operationsaufwands und der zu erwartenden Kosten wird das Medikament jedoch nicht für jeden verfügbar sein.
„Das wird sicher teuer“, sagt Prof. Wild.
Einen offiziellen Preis für das Medikament gibt es nicht. Gentherapien sind zwar oft teuer, können aber aufgrund ihrer langfristigen Wirkung dennoch erschwinglich sein. In Großbritannien übernimmt der NHS die Kosten für eine Gentherapie gegen Hämophilie B für 2,6 Millionen Pfund pro Patient.
Prof. Tabrizi sagt, diese Gentherapie sei „der Anfang“ und werde die Tore für Therapien öffnen, die mehr Menschen erreichen können.
Sie würdigte die „wirklich mutigen“ Freiwilligen, die an der Studie teilgenommen hatten, und sagte, sie freue sich „überglücklich für die Patienten und Familien“.
Sie arbeitet bereits mit einer Gruppe junger Menschen, die wissen, dass sie das Gen haben, aber noch keine Symptome aufweisen – bekannt als Huntington-Stadium Null – und möchte den ersten Präventionsversuch durchführen, um herauszufinden, ob die Krankheit deutlich verzögert oder sogar vollständig gestoppt werden kann.
BBC

